How Much Does Medical Device Development Cost?
What are medical devices?
Medical devices cover many nonpharmaceutical products used for disease treatment, monitoring, or prevention. For example, medical instrumentation (electronics), mechanical devices, and combination devices are all included in the definition of a medical device. Some combination devices have chemical, pharmaceutical, or biological components. Medical devices also cover implantable devices, devices used during surgical procedures, and non-implantable devices used at home by patients. With so many medical devices out on the market, medical device importance for hospitals and our healthcare industry cannot be understated. This article looks at what medical devices are, medical device development company benefits, medical device development processes, medical device application fees, and medical device cost for FDA approval.
Is my product considered a medical device by the United States Food & Drug Administration (FDA)?
All medical devices are FDA regulated. Examples of manufacturers that must list their devices with the FDA can be found HERE. Your product qualifies as a medical device based primarily on its’ intended use and indications for use. The Food, Drug, and Cosmetic Act defines a medical device as:
“An instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is: 1) recognized in the official National Formulary, or the United States Pharmacopoeia or any supplement to them, 2) intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or 3) intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes. The term “device” does not include software functions excluded pursuant to section 520(o).”
What are the FDA classifications for medical devices?
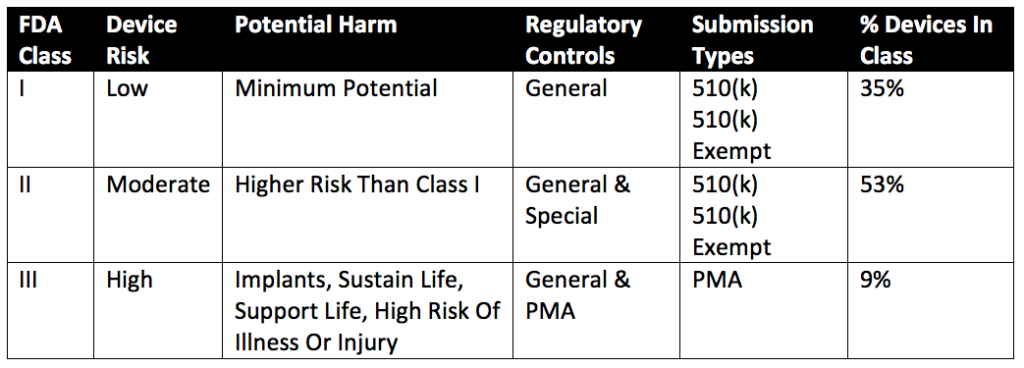
The FDA groups devices into one of three regulatory classes based on the potential risk devices have to human health during intended use. Class I devices present little to no potential harm to humans. Class II devices present more risk than Class I devices. Implantable devices, devices that support or sustain life, or devices with a high risk of illness or injury when used fall under Class III devices. Class I and II devices only require a 510(k) submission. Class III devices require a full premarket approval (PMA) submission for FDA marketing clearance. Some Class I and Class II devices fall under an exemption for 510(k) submissions. All exempt devices are subject to limitations on their exemptions. Information on the limitations of device exemptions can be found under 21 CFR Parts 862-892. Additionally, substantially equivalent Class III devices or Class III devices marketed before the FDA’s 1976 amendments (pre-amendment devices) only require a 510(k) submission instead of a complete PMA. Note that pre-amendment devices with “significant changes” will require a full PMA for marketing approval. In the case of Class III substantially equivalent devices, both the intended use of the device and the indications for use are considered for a 510(k) vs. a PMA submission. Further details on Class I, Class II, and Class III devices and the approximate percentage of medical devices developed by companies in each Class (either through their own medical device development processes or through outsourcing to a medical device development company) can be found in Table 1 below (reproduced from a presentation by Kimberly Piermatteo) and in our article on FDA medical device classifications.
How much does it cost to make a medical device?
The costs for getting a new medical device to market are various medical device development processes differs based on the type (Class I, Class II, or Class III) of medical device marketed and its intended use. Many factors influence the finances needed to fund a new medical device. Some of these factors include the types of medical device development processes required for the new device. For example, a medical device start-up may need more funds and to contact a medical device development company, contract testing company, and clinical studies coordinators if the novel device requires biocompatibility and quality control testing, needs clinical studies, and has a high degree of medical device technology complexity (or novelty).

In 2010 data was collected and published based on a survey sent to 750 of the 1,000 medical device companies in the industry. With data from the 200 companies that responded, the average total cost to get a Class II 510(k) product from concept to FDA clearance was $31 million dollars. Of this $31 million, approximately $24 million was spent on FDA-dependent or related tasks. Additionally, $2 million to $5 million dollars of the $31 million were spent on initial development and engineering costs. On average, engineering and development costs are 12% of the total cost of getting a 510(k) medical device to market. In cases where clinical studies are not needed, the $2-$5 million in development costs becomes closer to 30% of the total cost of the medical device project.
In contrast, the average total cost to get a Class III medical device from concept through premarket approval (PMA) was $94 million dollars. Of this $94 million, approximately $75 million was spent on FDA-dependent or related activities. Table 2 below provides a chart of the funding raised for various medical devices. As you might expect, medical devices that needed clinical trials (Class III devices) needed to raise significantly more capital. For those companies that have not acquired regulatory clearance, additional equity may be needed to address clinical findings, complete manufacturing transfers, and finalize scale-up.
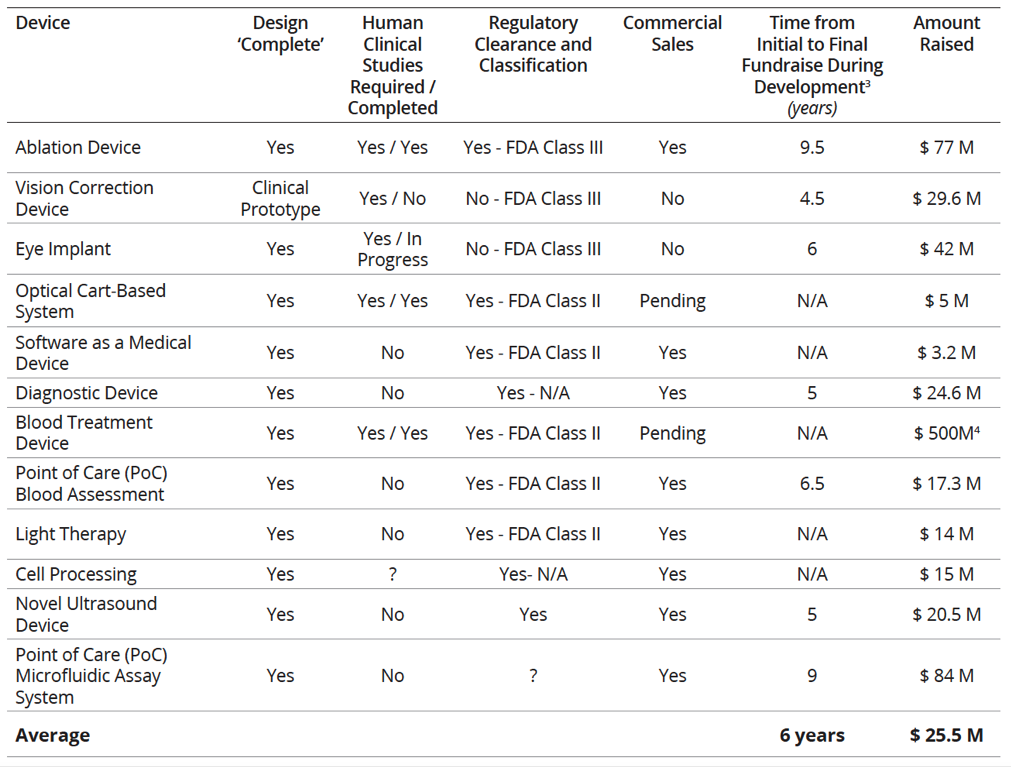
For the 2010 study, 510(k) clearance took an average of 20-months for initial development and proof of concept work, 12-months for clinical development, and 40-months to 510(k) clearance. All told, moving a 510(k) medical device from initial development through clearance was about 6-years. PMA medical device timelines were roughly double that of 510 (k) devices at 12-years.
How much do FDA fees for new medical device applications cost?
The current FDA application fees for medical devices can be found here.
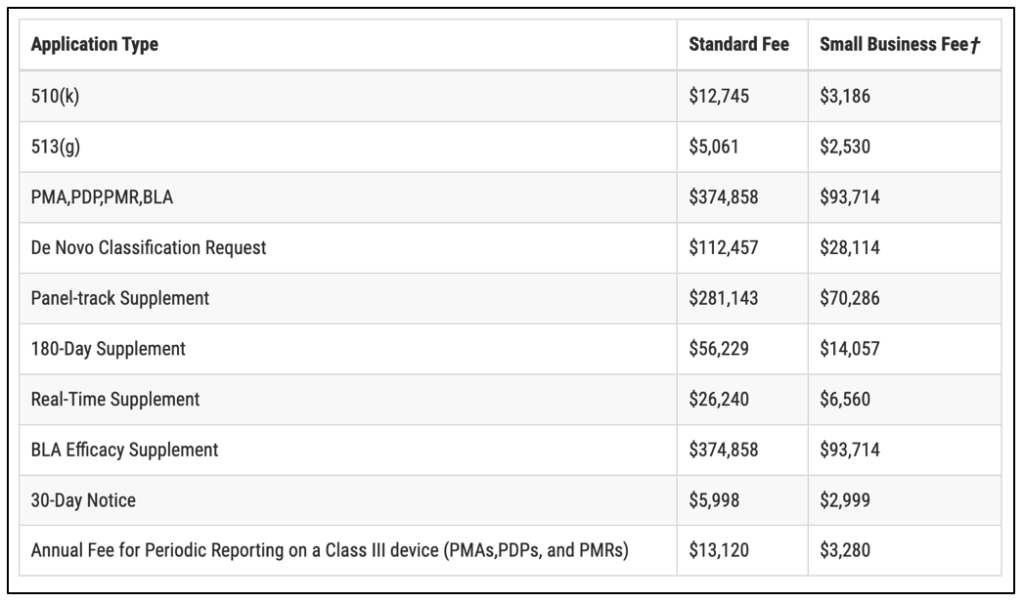
The FDA fee schedule for 2022 (updated Oct. 2021) has been reproduced in Table 3 below. You can assume that a startup will qualify for a small business fee. Note that businesses submitting small business fees should be certified by the Center for Devices and Radiological Health (CDRH) as a small business. Small business fees are generally half to a quarter of standard FDA fees.
Summary
Overall, medical devices cover a vast range of nonpharmaceutical products used for disease treatment, monitoring, or prevention. Some combination devices have chemical, pharmaceutical, or biological components. Medical devices also cover implantable devices, devices used during surgical procedures, and non-implantable devices used at home by patients. The FDA groups devices into one of three regulatory classes based on the potential risk devices have to human health during intended use. Class I devices present little to no potential harm to humans. Class II devices present more risk than Class I devices. Implantable devices, devices that support or sustain life, or devices with a high risk of illness or injury when used fall under Class III devices. Class I and II devices only require a 510(k) submission. Class III devices require a full premarket approval (PMA) submission for FDA marketing clearance. The average total cost to get a Class II 510(k) product from concept to FDA clearance was $31 million in 2010. In contrast, the average total cost to get a Class III medical device from concept through premarket approval (PMA) was $94 million dollars. Adjusting for inflation between ~2010 and 2020 and these values increase by approximately 20% to $37.2 million dollars for Class II 510(k) devices and $112.8 million dollars for Class III PMA devices. All told, moving a 510(k) medical device from initial development through clearance was about 6-years. PMA medical device timelines were roughly double that of 510 (k) devices at 12-years. All in all, whether you are beginning your medical device journey, or it is a couple of years down the road, ensure you choose a contract testing organization that can support you with appropriate biocompatibility testing for your medical device, implant, or product.
Ethide Labs is a contract testing organization that specializes in various biocompatibility and toxicity tests for medical devices, implants, and other products. Ethide Labs also offers Microbiology Testing, Bioburden Testing, Cytotoxicity Testing, Ethylene Oxide Residual Testing, Bacterial Endotoxin Testing, Sterility Testing, Environmental Monitoring & Package Integrity Testing services for medical device companies and allied industries. Ethide is an ISO 13485 certified facility.
References
Josh Makower, Aabed Meer, & Lyn Denend. FDA Impact of U.S. Medical Technology Innovation – A Survey of Over 200 Medical Technology Companies. 2010.
Mark Drlik. How Much Does it Cost to Develop a Medical Device? StarFish Medical Article. 2020.
United States Food & Drug Administration. How to Determine if Your Product is a Medical Device. United States FDA Article. 2019.
United States Food & Drug Administration. Medical Device User Fee Amendments (MDUFA). United States FDA Article. 2021.
Share this in your social networks